Study Reveals Key Mechanisms of a Rare Form of Epilepsy
April 15, 2024
Article published by MedicalXpress
*Featuring work by CURE Epilepsy Grantees, Drs. Edward Cooper and Jeffrey Noebels
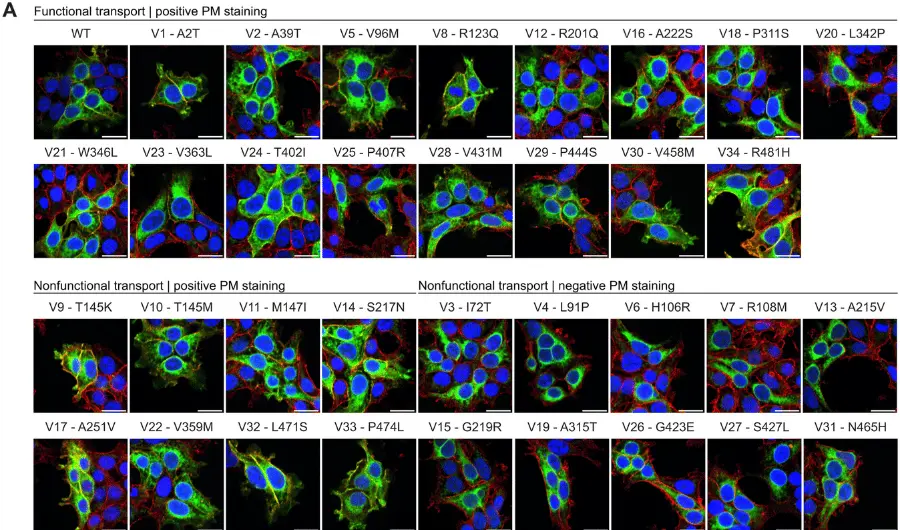
New research is advancing the understanding of KCNQ2 encephalopathy. “KCNQ2 encephalopathy is a rare neurodevelopmental disorder caused by variants in the KCNQ2 gene, which provides the recipe for a type of brain potassium channel,” explains lead author Timothy Abreo, a student in the Genetics and Genomics Graduate Program at Baylor College of Medicine, Houston, Texas, US. “The disorder usually manifests as seizures beginning within days after birth and developmental delays which are lifelong and without effective current treatment.” Newly identified variants in the KCNQ2 gene are hard to assess because not every variant is disease-causing, and the reasons some variants lead to illness have been poorly understood. The research team closely examined a specific genetic variant of the KCNQ2 gene involving a single amino acid change—glycine 256 changed to tryptophan (G256W)—on one copy of the KCNQ2 gene. They examined the structure and pathogenicity of the G256W variant using molecular, cellular, and in vivo approaches and uncovered evidence that G256 helps form a network of hydrogen bonds that serve to stabilize the structure of the pore-forming region of the potassium channel. “Taken together, our study reveals that the presence of KCNQ2 G256W variants affects both molecular and cellular aspects of KCNQ channel activity, including their ion-carrying capacity, expression, and placement within cells,” explains senior author Edward Cooper, Associate Professor of Neurology, Neuroscience, and Molecular and Human Genetics at Baylor College of Medicine. “Selective KCNQ2 openers, which counter the effects of G256W mutations should be tested in additional laboratory models and if effective, in humans with KCNQ2 disease-causing variants,” says Dr. Cooper.
Latest from the Community